
Are we getting closer to curing Alzheimer’s Disease?
Are we getting closer to curing Alzheimer’s Disease?
---- Beta-amyloid and beyond

On January 6, 2023, FDA approved a new drug for Alzheimer’s Disease (AD), Leqembi (previously known as lecanemab). This is the second FDA-approved AD drug targeting the biological roots of the disease pathology, instead of temporarily relieving the symptoms. Leqembi is now commercialized by Eisai at a price tag of $26, 500 for a one-year treatment. About 6 million Americans suffer from AD. This drug has the potential of becoming a blockbuster, even though Eisai is being conservative on the sales projection now as the reimbursement decision by Medicare will not come out until later this year.
Lecanemab is a monoclonal antibody (mAb) targeting beta-amyloid (Aβ), a protein that aggregates in the brain of AD patients. Yet not a cure for AD, Leqembi can buy some time for early-stage AD patients by delaying cognition decline and memory loss. The success of Leqembi provided evidence supporting the Aβ theory, which the field is drifting away from after the repeated failures of Aβ-targeting agents in clinical trials.
Does it mean we are getting closer to curing AD?
To shed light on this question, I will dive into AD therapeutic development. In this post, I will focus on the beta-amyloid hypothesis and what makes lecanemab the best Aβ targeting therapy so far. In the next post, I will discuss alternative pathways and drug targets, promising therapeutics, and how we can leverage the current techbio and digital bio platforms to facilitate neurodegenerative drug discovery.
The beta-amyloid theory
AD has been identified over a century ago, but the biological mechanism that causes the disease is still unclear. Among all attempted explanations, the Aβ hypothesis is well supported by neuropathological and genetic data and has become the most received theory.
Aβ is a peptide derived from the amyloid precursor protein (APP), a receptor on the surface of neurons that regulates synapse formation and neuronal plasticity. Cellular enzymes such as β- and γ-secretases recognize multiple cutting sites in APP and digest APP into Aβ peptides with various lengths. The Aβ monomers are not toxic in healthy brains when their production and clearance are in equilibrium. However, once the clearance is disrupted due to aging or other AD risk factors, over-produced Aβ monomers accumulate and aggregate into oligomers and eventually form dense, insoluble plaques.
Aβ hypothesis posits that the aggregation of Aβ proteins in the brain is the central event that leads to neurodegeneration and AD pathology. However, accumulating data suggest that it is not Aβ but tau deposition that is directly linked to neuronal death and cognitive deterioration. Instead, Aβ aggregates were proposed to be the ‘threshold archiver’ that gates downstream neurotoxic signaling cascade. When the Aβ load is below the threshold, tau deposits are locally expressed and considered benign. As the brain Aβ concentration increases and the amyloid burden goes above the threshold, tau pathology is intensified and spread across multiple brain regions, which leads to global neuronal loss and cognition deterioration.
Based on this theory, reducing the brain Aβ to the subthreshold level is expected to halt or delay the disease progression in AD patients. The intrinsic complexity in AD biology and the heterogeneous brain Aβ composition make Aβ a challenging target for therapeutic development. A few questions need to be addressed to develop effective anti-Aβ therapies: 1) Which Aβ species(s) is (are) the most relevant to neurotoxicity? 2) Where is the ceiling of the therapeutic effects for Aβ-targeting agents? 3) What is the best dosing timeframe for AD patients to achieve the maximal clinical benefits?
Amyloid targeting agents
During the past two decades, over 40 therapeutic agents that were developed to restore Aβ homeostasis have reached clinical stages, but over 95% of these clinical trials failed. Among all the Aβ-targeting therapeutics, three classes of strategies have been employed.
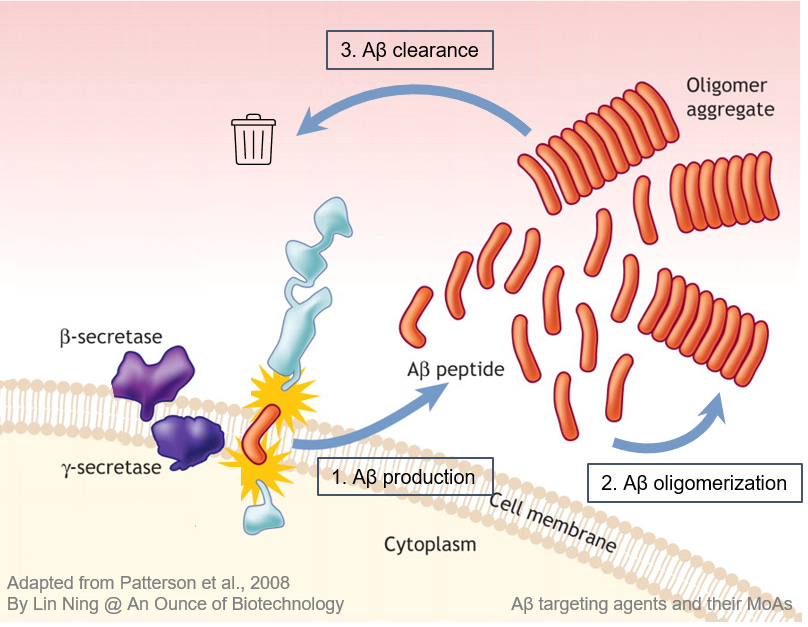
Preventing Aβ production
Inhibition of enzymes that process APP into Aβ can slow down Aβ production and prevent its aggregation. Inhibitors targeting BACE-1 and γ-secretases have reached late-stage clinical trials. However, these agents all failed before or at phase III due to a lack of efficacy or safety issues, owing largely to the off-target effect on other signaling pathways that are fundamental for healthy brain functions.
Blocking Aβ oligomerization
Antibodies and small molecule drugs bind to Aβ monomers and block their binding site for other Aβ monomers and therefore prevent Aβ aggregation. Among them, ALZ801 is the most advanced program now being tested in a Phase III trial. ALZ801 has shown promising clinical efficacy in a Phase II study suggesting blocking Aβ oligomerization is a viable strategy. Other programs are still in the preclinical or early clinical stages.
Facilitating clearance of existing Aβ aggregates
Anti-Aβ immunotherapies were employed to remove existing Aβ aggregates by activating microglia or other immune cells to enhance the phagocytosis of Aβ proteins. Among these agents, anti-Aβ monoclonal antibodies (mAbs) have been extensively studied. Currently, several anti-Aβ mAbs have reached late clinical development with favorable features and promising clinical outcomes.
Anti-Aß monoclonal antibodies
Here I will review seven late-stage anti-Aβ mAbs, including bapineuzumab, solanezumab, crenezumab, donanemab, gantenerumab, and the FDA-approved aducanumab and lecanemab, on their Aβ binding selectivity, safety profiles, and clinical outcomes.

Non-selective anti-Aβ mAbs
Bapineuzumab and Crenezumab were designed to non-selectively remove Aβ at all aggregation states. In vitro binding assays confirmed their high affinity to Aβ plaques and soluble oligomers and moderate affinity to the monomers.
In two phase III trials, in which patients carried high levels of amyloid burden at the baseline, bapineuzumab managed to keep the Aβ plaque levels unchanged during the course of treatment, while Aß deposits increased rapidly in the placebo-controlled arm. Interestingly, bapineuzumab significantly reduced the CSF p-tau level, indicating regulation of tau hyperphosphorylation is not correlated with Aβ plaque load. Crenezumab also failed to induce Aβ plaque removal in a phase II trial. However, it decreased the CSF Aβ oligomers by ~50% while increasing the levels of Aβ monomers, suggesting the successful target engagement with Aβ oligomers and blockade of new oligomer formation from monomers. Crenezumab showed no effect on p-tau and t-tau levels in CSF.
In mild-moderate AD patients, bapineuzumab and crenezumab failed to show clinical efficacy in preventing cognitive decline at their highest tolerated dose, as measured by ADAS-Cog and CDR-SB. As for the safety profiles, up to 15% of bapineuzumab-treated patients developed ARIA (Amyloid related imaging abnormalities), a common side effect due to Aβ plaque removal, while crenezumab did not induce ARIA. Given their high affinity to Aβ plaques, the inefficient plaque clearance may be a result of antibody saturation with soluble Aβ and low target engagement in plaques. Bapineuzumab’s failure in Aβ plaque clearance may be partially due to more severe Aβ pathology at baseline, as evidenced by the high amyloid burden and the rapid worsening in the placebo arm. The failure of crenezumab also suggests that removing oligomeric Aβ does not necessarily lead to clinical benefits for AD patients.
Aβ monomer-targeting mAb
Solanemzumab was developed to target soluble Aβ monomers and oligomers, following the peripheral sink hypothesis, which posits that the Aβ plaques serve as the reservoir; lowering the peripheral Aβ levels promotes the release of Aβ from the reservoir and disaggregates of the plaques. In vitro binding assays confirm its strong affinity to Aβ monomers and oligomers, but not plaques.
In three phase III trials, solanemzumab showed successful target engagement and induced a shift in brain Aβ equilibrium from the insoluble to the soluble pool, but failed to reduce the brain Aβ plaques significantly. Solanemzumab also showed no effect on p-tau and t-tau levels in CSF.
In these trials, solanemzumab exhibited no clinical efficacy with the exception of a small effect in ADAS-ADL in a subgroup of patients with milder symptoms. These results suggest that the removal of soluble Aβ monomers does not induce sufficient disintegration of Aβ plaques and has no clinical benefit.

Aβ plaque-targeting mAbs
Gantenerumab, aducanumab, and donanemab are all plaque-targeting anti-Aβ mAbs, with subnanomolar affinities to insoluble Aβ plaques. Donanemab is considered to be plaque-exclusive. Aducanumab and gantenerumab both bind to Aβ oligomers with different affinities (aducanumab KD=4-138nM, gantenerumab KD=4nM). Notably, gantenerumab also showed weak binding to Aβ monomers.
When dosing at 105mg and 225mg every 4 weeks in prodromal AD patients, gantenerumab did not induce a significant reduction of Aβ plaques in the brain, but a significant reduction in p-tau and t-tau in the CSF. When dosing up to 1200mg in three open-labeled trials, a 66-78% reduction of Aβ plaques was detected and over 50% of patients turned amyloid-negative (brain Aβ PET signal below 24 centiloid) by the end of 24-month treatment! Despite the sufficient plaque removal, gantenerumab seemed not very effective in improving cognitive functions, as the treated patients showed a faster cognitive decline compared to those in the trials of aducanumab, donanemab, and lecanemab. As reported in November 2022, in the latest phase III GRADUATE trials, 1200mg gantenerumab cleared only half as much plaque as expected and did not generate expected clinical benefits after 2-year treatment. In addition, this dosage is associated with a high rate of ARIA, close to 25%. These results led to the discontinuation of gantenerumab.
Aducanumab was developed from the serum components of the elderly that are resistant to developing dementia. The resultant mAb shows high affinity to Aβ plaques, moderate affinity to the oligomers, and no binding to the monomers. In two parallel phase III trials EMERGE and ENGAGE, aducanumab showed a 62% and 51% reduction in Aβ plaques respectively at 18 months. In addition, significant reductions in tau deposits as well as CSF p-tau and t-tau were also detected. Unfortunately, the reduction in amyloid burden and tau pathology did not lead to consistent clinical improvement. In the EMERGE trial, aducanumab induced a 22% slower reduction in CDR-SB at the high dose compared to the placebo arm, while showing no significant improvement in the ENGAGE trial. Despite the contradictory results and inconclusive clinical efficacy, aducanumab was approved by FDA in June 2021 (commercial name Aduhelm) and became the first disease-modifying AD drug.
Donanemab was developed as a plaque-specific mAb that binds to Aβ p3-42, a modified Aß peptide with the first two residues trimmed from the N-terminus and a pyrol ring formed at the third residue. Due to its aggressive aggregation nature, Aβ p3-42 is only present in the plaques and protofibrils. In vitro experiments showed its strong affinity to Aβ plaques. As expected, donanemab performs much better in plaque clearance. In the phase II Trailblazer-ALZ trial, 1400mg monthly treatment of donanemab reduced amyloid plaques by 64% at 6 months and 79% at 18 months, and over 60% of patients turned Aβ-negative. Importantly, the active arm patients were switched to placebos once they turned Aβ-negative, and the plaque levels remained low after the treatment was stopped. In this trial, Eli Lilly used integrated ADRS (iADRS) as the primary endpoint to measure cognitive functions, as they believed that this measurement is more consistent and sensitive. The active arm showed a 31% slower decline in cognitive functions than the placebo arm by the end of the 18-month trial. This is the first time an anti-Aβ mAb showed statistically significant benefit in phase II clinical trial. Three phase III clinical trials are ongoing testing donanemab in larger populations, and the results are expected by 2024. In October 2022, an interim analysis of the phase III Trailblazer-ALZ 4 confirmed donanemab’s superior capability in plaque clearance when compared side by side with aducanumab.
Aβ protofibril-targeting mAb
Lecanemab was developed to target the protofibrils, a type of soluble, high-molecular Aβ oligomers that are believed to be more toxic than Aβ plaques and might be the real culprit of AD pathology. Bioarctic, a Swedish biotech company found out that Aβ with the arctic mutation (E693G) is highly prone to form protofibrils. Therefore, they immunized mice with this mutated Aβ42 and identified a mAb m158, which was further humanized to lecanemab. This antibody has strong structural specificity to the epitopes in protofibrils. When compared to aducanumab side by side, lecanemab has an 80-105 folds higher affinity to protofibrils. Lecanemab also binds to the Aβ plaques with a nanomolar range affinity. In the phase III CALRITY-AD trial, when dosed on patients with mild cognitive impairment at 10mg/kg bi-weekly, lecanemab already showed significant amyloid clearance after 3 months and the reduction of the plaques reached 72% by 18 months. In addition, tau deposits and CSF p-tau and t-tau were all significantly reduced between 12-18 months. More importantly, significant improvements in CDR-SB (26.7%) and ADAS-Cog (27.2%) were observed after 6 months. The adverse events are much lower than other plaque-targeting mAbs, with 12.6% ARIA-E and 17.3% ARIA-H. The superior clinical efficacy and safety profiles made lecanemab the best AD drug out there.
The ceiling of the plaque-removal mAbs
Among all the anti-Aβ mAbs reviewed above, four mAbs effectively removed Aβ plaques: Gantenerumab showed the highest total plaque clearance (ΔAβ=-90.3CL and -74.9CL), followed by donanemab (ΔAβ=-84.5CL), lecanemab (ΔAβ=-58CL), and aducanumab (ΔAβ=-31.4 CL and -27.4CL). The plaque clearance levels were pretty impressive, which led us to expect a corresponding improvement in cognitive functions. However, only 3 out of 4 mAbs showed efficacies in delaying the deterioration of cognitive function, with the exception of gantenerumab. Notably, gantenerumab took 36 months to achieve that level of Aβ clearance while others took only 18 months. When I normalized the plaque clearance level to the time, donanemab exhibits a plaque clearance rate of 11.3CL per month, lecanemab at 5.67CL per month, and gantenerumab at 3.5 and 3.9 CL per month. A lower plaque clearance rate may contribute to gantenerumab’s failure in achieving clinical efficacy.
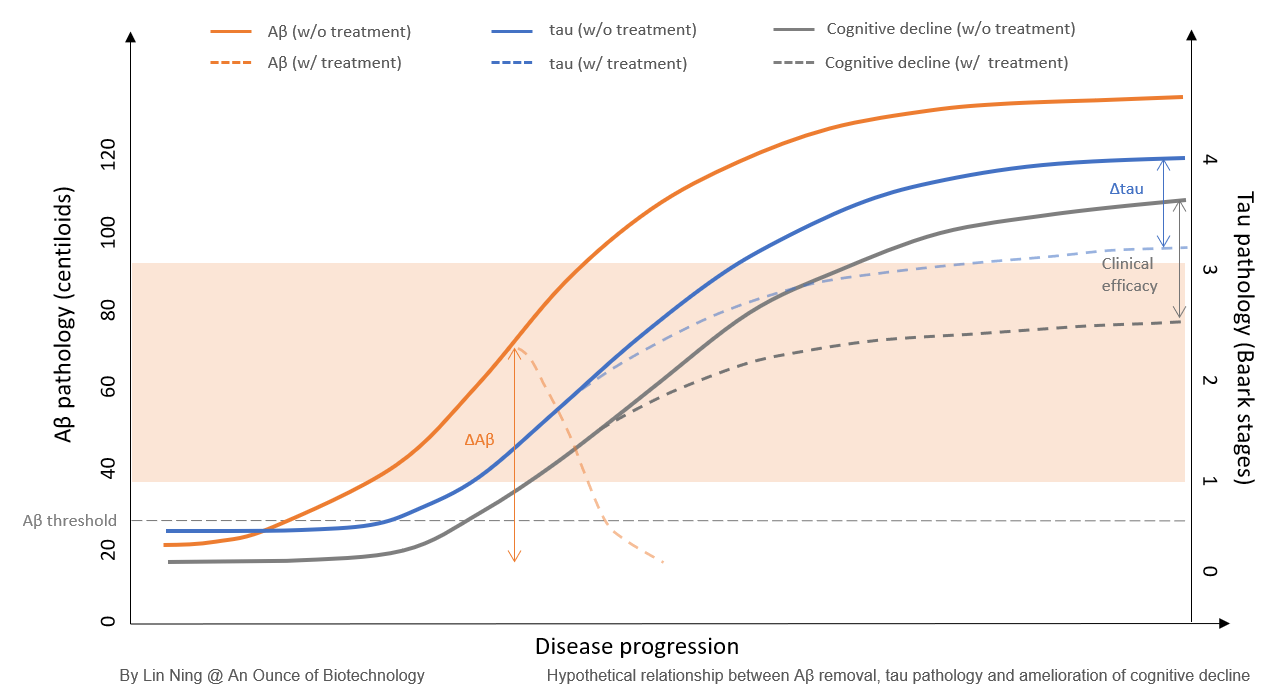
These data support the ‘Aβ threshold’ model. For any amyloid targeting agents, their ability to rapidly bring the amyloid burden to a subthreshold level is crucial to achieving the expected clinical efficacy, as the disease deterioration continues when the Aβ load is above the threshold even if some Aβ plaques are being removed. If it takes too long to reach the subthreshold Aβ level, the disease could have already progressed to a more advanced stage with uncontrollable, self-propagating tau pathology.
Can we further improve the efficacy of anti-Aβ plaque mAbs by increasing their Aβ clearance rate? Assuming it is feasible to increase the Aβ clearance rate by optimizing affinity and target engagement, rapid plaque removal might put the patients at risk of developing ARIA, as a high incidence of ARIA is associated with all Aβ plaque-targeting mAbs but not with those targeting monomeric or oligomeric Aβ. The Aβ clearance rate and the safety profile must be balanced when developing Aβ plaque-targeting mAbs, or the adverse effects will set a major limitation on the therapeutic window. Donanemab might be the ceiling of Aβ plaque-targeting mAbs and a ~30% clinical efficacy is probably the best this class of therapeutic agents can achieve.
How about the oligomers?
Accumulating evidence from preclinical studies supports the role of soluble, oligomeric Aβ in mediating neuronal death and synaptic neurotransmission. This is an appealing hypothesis, as it nearly resolves the issues regarding the lack of correlation between Aβ plaque levels, tau pathology, and cognitive decline. Oligomeric Aβ might cause direct toxicity to neurons that result in synapse loss and neuronal death. However, this remains a highly complex area of science and requires supporting data from clinical trials.
As the first oligomeric Aβ-targeting mAb that marked all the biomarker endpoints and achieved clinical efficacy, lecanemab is expected to provide insights into how Aβ oligomers, especially protofibrils, are involved in AD pathogenesis. Since lecanemab also binds to the Aβ plaques with medium-high affinity, the clinical benefits might be a mixed effect of removing both Aß protofibrils and plaques. Lecanemab showed clinical benefits as early as 6 months into the treatment, while donanemab and aducanumab did not show clinical efficacy until 18 months. In addition, lecanemab exhibited a much lower ARIA rate at 12.6%, in contrast to 27.5% with donanemab and 35% with aducanumab. Moreover, lecanemab induced a significant reduction in the levels of tau pathology, glial cell activation, and synaptic dysfunction as early as 6 months, indicating a direct effect in correcting errors in glial signaling and synaptic toxicity. These different behaviors of lecanemab suggest its clinical benefits are at least partially attributed to protofibril removal. The therapeutic effect of targeting Aβ oligomers is likely to flatten the curve of tau pathology and cognitive decline during early pathogenesis, independent from the Aβ plaque threshold level. New methods detecting protofibrils are required to quantitatively measure changes in protofibrils in the brain and CSF to evaluate the target engagement and therapeutic mechanism of lecanemab.
Notably, ALZ-801 (valiltramiprosate), a brain-penetrant small molecule drug, was developed by Alzheon to block the oligomerization site in Aß monomers and prevent aggregation but does not trigger plaque clearance. In two parallel phase II studies, ALZ-801 induced clinical improvement over placebo of 40-66% and 25-45% on ADAS-Cog and CDR-SB respectively in APOE4 homozygotes with mild AD symptoms. These results are still to be confirmed with a large patient population, but the preliminary data in the phase II trial confirmed that high molecular-weight Aß oligomers are the favorable AD targets.
The best timing for amyloid beta therapeutics to work
Selecting the right patient population for treatment plays a critical role in the success of clinical trials. Clinical scores are the most used matrix to define patients within a targeted disease stage. For AD trials, the clinical outcomes are better when the intervention was applied to patients with mild symptoms. Biomarkers are also employed to further enrich the patient population for better efficacy. This is particularly important for AD trials because changes in biomarkers occur way ahead of clinical symptoms and amyloid deposition is poorly correlated with tau pathology and cognitive decline.
For Aβ plaque-targeting therapeutic agents, the ideal timing to start the treatment can be mapped out by the Aβ and tau pathology levels:
The amyloid-positive state
Several human studies showed that the elderly with Aβ burden below 30CL is generally free from AD pathology and cognitive impairment. Therefore, the industry has been using 20-25CL as the Aβ threshold, above which Aβ plaques can potentially trigger downstream pathological events. For any anti-Aβ therapeutic agents to be effective, the brain Aβ load must be higher than the threshold level.
Tau pathology positive but not yet self-propagating
The Baark scale has been proposed to model tau pathology and its relationship with cognitive decline has been established. There are 5 Baark stages based on the tau PET SUVR values. Stage 0 (0-1.129) is considered to be free of tau pathology and signs of cognitive abnormalities. Stage 1-2 (1.129-1.523) is when tau pathology initiates and spreads from the media temporal lobe to other brain regions and memory and cognitive declines start. At these stages, tau pathology alone is not sufficient to develop cognition decline and amyloid pathology is required. Stage 3-4 (>1.523) is when tau pathology becomes widely spread and self-propagating. Cognitive deterioration occurs without amyloid. Therefore, stage 1-2 makes an ideal target for anti-Aβ mAbs. In donanemab Trailblazer-ALZ trials, tau PET SUVR between 1.1-1.46 was one of the inclusion criteria for patient selection, which falls into the Baark Stage 1-2.
For Aβ oligomer-targeting therapeutic agents, there is not enough clinical data to build a model to illustrate the interplay between Aβ oligomers, tau pathology, and cognitive functions. If the Aβ oligomers directly drive tau pathology, lowering the levels of Aβ oligomers might decrease the probability to develop tau pathology at an early stage and postpone the disease onset.
Is beta-amyloid still a promising AD target?
If a drug target fits the deterministic model to a disease, treatment at the pre-symptomatic stage would interrupt the disease process and prevent clinical manifestations, whereas treatment at the symptomatic stage would be expected to at least halt clinical deterioration, especially in the presence of good target engagement.
Imatinib, a small molecule inhibitor of BCR-ABL tyrosine kinase, indeed halted clinical deterioration in chronic myeloid leukemia. Almost all CML patients respond to imatinib and treatment with imatinib is necessary and sufficient to treat chronic myeloid leukemia at any disease stage.
The clinical data from recent anti-Aβ agents trials suggest an alternative model: Aβ is still a key factor in AD pathophysiology, but it is not the only cause that drives the AD cascade and is not proportional to the clinical symptoms. Anti-Aβ agents can be meaningful for the patients by slowing down cognitive deterioration by ~ 30%. However, they are not going to be the silver bullet that halts the disease progression. Alternative pathways and drug targets have been explored and some of them are showing promising results, which I will cover in the next post.
This an excellent summation of the drug development and efficacy of amyloid plaque targeting therapeutics. The questions is still whether these plaques are casual to AD...